
TIFFANY HOUSE
December 2004
Several months ago, I was given the opportunity to write a paper about an experience that changed my life. I wrote about being diagnosed with Pompe’s disease.
I found that writing about my experiences was a good experience for me.
To read this paper, please click here.
Tiffany
Age 21
1999
I was born on January 22, 1983. By the time I was 3 months, I had had my first cold. I developed slowly for my age, but not slowly enough that doctors noticed that something was wrong. As a little kid, I fell a lot. I always seemed to have a skinned knee. I would also always catch a cold during Christmas or January, my birthday. It never failed. Do you have any idea what is like to be sick at the same time every year? When I was 7 I had pneumonia for the first time. I was so sick that I missed over a month of school.
Ever since I was little, I have been going to a wide variety of doctors. I have never been able to bend over. My back just doesn’t curve like it is supposed to. When my mom would ask doctors, they would say, “Maybe it is normal for her.” She even thought at one point that I might have Cystic Fibrosis, so she had me tested. When the test came back negative, she was relieved. Meanwhile, I had no idea anything was wrong. I thought that I was just like everybody else. This went on until I was 11 years old. About a week before Christmas that year, my dad’s cousin, who is a pediatrician at the Mayo Clinic, was in town for a convention. My parents told him about my medical history, and asked him what he thought. I remember him asking me to walk up a flight of stairs, and to walk down a hall. Within 10 minutes he told my parents that he thought that I had a myopathy. That was all it took. I had been going to doctors for years that said that nothing was wrong, and he diagnosed me in 10 minutes by just watching me! He arranged for me to have a muscle biopsy done at the Mayo Clinic in January. I was diagnosed the week before my 12th birthday. At the time I was 4 feet 10 inches and weighed only 58 pounds. After the biopsy, I went through a wide variety of tests. We found out that I also had very bad respiratory problems, my lung functions were only 40% of normal. The doctors told me that I would only live into my second decade. They also told us that there wasn’t anything we could do about it.
The week after I got back, I was put onto a respiratory machine called the Bi-Pap. It was really hard going back to school and knowing that I was really sick, when I didn’t feel sick. For the next year, things started to get harder, but I was still able to go to school. But by the spring of ’95, I started missing a lot of school until I was no longer able to attend. The next year I started homeschooling. After that things started to get bad fast. By June of ’97 I had developed scoliosis, and my lung functions were below 20%. The good news was that I had grown and gained weight. Unfortunately, as I gained weight, my scoliosis increased. Right now, I weigh 90 pounds and am 5 feet 3 inches. My scoliosis is now over 90%. In March of this year (1999—age 16) I had to start using a wheelchair. It was one of the hardest decisions that I have ever made, but I didn’t have a choice.
I think one of the hardest aspects of having this disease has been missing out on so much of my adolescence. Sometimes when I think of everything that I have missed out on, I get really depressed. I mean, I missed out on going to my eighth grade graduation, lots of school dances, dating, learning to drive, and countless other things. It is also really hard to accept it when things that I used to be able to do, I can’t do anymore.
There are so many people of all ages out there with this disease. I know a little girl who has the infantile form. The first thing that tipped the doctors off that something was wrong, was her enlarged heart. Instead of trying to find out why, they told her parents that they would find out during her autopsy. She is just one of so many, and none of them are expected to live past a year. There are also adults out there that have it. Some of them were even diagnosed as teenagers. I have gone through many changes since I was diagnosed almost 5 years ago. Most have been bad, but I’ve also formed some wonderful friendships with people who have taken an interest in me and have tried to help. Thank you for reading my story.
Tiffany
Age 16
June 1999—Juvenile Clinical Trials in Rotterdam, the Netherlands
Tiffany was accepted into the first juvenile clinical trialsA research study that tests new treatments or approaches in people. with enzyme replacement therapy (ERT)A treatment that replaces the missing enzyme through IV infusion. in the Netherlands in June 1999. There she was treated with enzymeA protein that helps the body carry out chemical reactions. derived from the milk of transgenic rabbits. After a year of treatment at Sophia Children’s Hospital-Rotterdam, she returned to the US to undergo back surgery for severe scoliosis (July 2000), at the Mayo Clinic in Rochester, Minnesota. She continued to receive treatment at the Mayo Clinic until she was granted permission to receive treatment in San Antonio, Texas, her home (April 2001). She currently receives treatment at The University of Texas Health Science Center at San Antonio. She continued on the transgenic “rabbit” enzyme until July 2002, when she was transitioned to a CHO derived enzyme. She is currently the only delayed onset patient in the US in a clinical trial for Pompe’s disease.
July 2002—Transition to CHO Enzyme
After months of reduced supply of the transgenic product, Genzyme Corporation decided to cease production of the transgenic enzyme and to focus production on the CHO derived product. Tiffany was the first patient transitioned from the transgenic “rabbit” enzyme to the CHO enzyme in July 2002.
Progress on the Transgenic “Rabbit” Enzyme—June 1999 to June 2002:
During her 3 years on the transgenic enzyme, improvement in her condition seemed to occur primarily during her final year of treatment. This could be attributed to several reasons: (1) increase in dosage; (2) recovery from back surgery.
In the Netherlands the dosage in the juvenile patients was minimal at onset of treatment. Her dosage was increased just 2 months prior to her leaving the Netherlands to undergo scoliosis surgery at the Mayo Clinic in Minnesota. She remained on the increased dose of transgenic enzyme for two years. During the first year, it was difficult to assess improvement in her physical condition because she was recovering from critical back surgery. After that, however, Tiffany began to show definite signs of improvement. Her stamina and strength increased; she felt better overall; her appetite increased; she had more energy and had very little muscle pain, nausea, and diarrhea. The most encouraging sign, however, was noted just prior to her transition to CHO. In June 2002, her pulmonary functions showed an dramatic increase. This was just prior to her being transitioned to the CHO enzyme. The first pulmonary evaluations after the transition showed a marked decrease in her pulmonary functions (to the lowest levels recorded in, at least, a year and a half).
November 2002—Tiffany’s Progress on the CHO Enzyme
Tiffany’s first infusionA method of delivering medication through an IV. (July 2002) with the CHO enzyme was relatively innocuous with only minor reactions noted. However, after four months of treatment with CHO, it is evident that Tiffany is once again experiencing problems that had previously subsided or diminished during her first three years of treatment with the transgenic enzyme. The problems that have re-surfaced include: urinary tract infections, nausea, diarrhea, upset stomach, muscle pain, headache, fatigueA strong sense of tiredness or low energy that does not fully improve with rest., loss of appetite, and weight loss. These are symptoms that developed gradually over the course of the disease and worsened as the disease progressed until they became chronic and monumental obstacles that plagued Tiffany on a daily basis.
Tiffany has been on ERT (in a clinical trial for enzyme replacement therapy) for three years and four months (3 years on the transgenic “rabbit” enzyme and 4 months on the CHO enzyme). This has impacted Tiffany’s life not only physically but psychologically as well. Although participation in a clinical trial offers hope, it is a virtual roller coaster of emotional experiences that are extremely monumental during teenage years. She was finally beginning to see a light at the end of the tunnel and beginning to notice improvement (after 3 years of treatment with the transgenic enzyme) when Genzyme required her to transition (as they will all of the transgenic patients) to the CHO enzyme and reduced her dosage by half.
Last year Tiffany entered college in spite of reservations about her ability to physically handle the daily routine. Prior to this she had not attended school in over 5 years. Tiffany has now completed her freshman year at the University of Texas at San Antonio. She is in the honors program and is on the dean’s list. This is a monumental step forward for someone that didn’t receive a high school diploma. She took the GED and scored well enough on the SATs to be accepted by all universities in the San Antonio area.
Quality of life issues are the main concern of the patient suffering from a debilitating and life threatening disease. Although examination of the clinical data is vitally important, personal contact with the patient and the patient’s own assessment of the treatment is also of great importance. Both objective and subjective data need to be examined closely. A patient’s input should be taken into consideration when making a judgment about the efficacy of a treatment. This is why trials in delayed onset patients are so important. The patient is able to give his reactions to the treatment. This is an invaluable tool in the development of any therapy.
Out of the four patients transitioned from transgenic to CHO in July 2002, three patients are having similar experiences to Tiffany’s. Although these changes could also be attributed to a variety of causes, it is also reasonable to attribute the reoccurrence of disease symptoms to an insufficiency in dosage of the CHO enzyme.
Although these issues have been brought up with Genzyme and the FDA, it remains to be seen how they will react to this digression in the condition of these clinical trial participants.
Following is a letter issued by Marylyn House, Tiffany’s mother, in order to bring attention to Tiffany’s current condition:
……. I appreciate your interest in Tiffany’s progress. However, I think that you misunderstood the point of my previous letter. I do not hold any hopes for Tiffany to resume treatment with the transgenic enzyme (although that is what we wanted). (At this point) What I want is for Tiffany to receive a higher dosage of the current enzyme.
When she was transitioned to the CHO enzyme, the dosage was reduced by half. At that time Genzyme claimed that the CHO enzyme was (at least) twice as effective as the transgenic product…….. The data that they used to support this theory was comparative analysis of the two products done “in house” by Genzyme. They denied access to the two enzymes by an outside party with which to conduct comparative analysis.
………(perhaps) the CHO product is NOT twice as effective……….and our greatest fear is surfacing. Tiffany is regressing!
Although we did originally want Genzyme to continue to supply the transgenic enzyme, we knew that when this treatment ceased, the opportunity to return to the transgenic enzyme was nil. Transgenic enzyme (at least from rabbit’s milk) will never be reinstated by Genzyme……… Although this is understandable from a business point of view, it is………(not) from a patient’s point of view………
Marylyn House
November 2002
UPDATE—DECEMBER 2002
A change in Tiffany’s infusion protocol was authorized by Genzyme in November to try to alleviate problems that she has encountered since her transition (and reduction in dosage) to the CHO enzyme in July 2002. It is still too early to tell if the change will be beneficial.
UPDATE—JANUARY 2004
When she was diagnosed at the age of 12, we were were told that Tiffany would not live to the age of 20. She has now reached a milestone. In January 2004, Tiffany celebrated her 2lst birthday.
At the age of 16, Tiffany was admitted as the first juvenile patient in the first clinical trials with enzyme replacement therapy at Sophia Children’s Hospital in Rotterdam, The Netherlands.
Many changes have taken place in the past 4 1/2 years and along this path, Tiffany has experienced many ups and downs. Currently, she shows improvement in her strength and stamina, but her respiratory functions remain critically impaired.
In most cases, dosage plays a key role in a patient’s road to improvement, and hence, dosage studies are part of Genzyme’s ongoing studies. Although Genzyme did implement a dosage increase in all of the original transgenic patients (all of whom experienced a decline after transitioning to a decreased dosage of CHO enzyme), Tiffany’s dosage is still below that received on the transgenic enzyme. Hopefully, she will receive a dose increase in 2004 to try to improve her respiratory functions which remain a critical aspect of this disease.
UPDATE—JUNE 2004
As of June 2004 Tiffany is now receiving the same dosage that she was receiving while on the transgenic enzyme.













Rachel’s Story
May 2026 The following story was written by Courtney, Rachel's daughter, and submitted to us to share her mother's experience. We are grateful for her willingness to give us this window into their family's journey. While we strive to share accurate and helpful...

Dwayne Update: 3-4-2025
Updated—March 2025 Hi everyone, my name is Dwayne; I am 57 years old. I was diagnosed with Late Onset Pompe diseaseA form of Pompe disease that begins after infancy and usually progresses more slowly. (LOPD) in November 2018 when I was 50 years old. I live in Southern California in the City of Irvine with my wife and mother-in-law. I am a father to...

McKenna’s Story
January 2022 I am McKenna Wellner and I was diagnosed with Pompe diseaseA rare genetic disease in which the body cannot properly break down glycogen, leading to buildup that damages muscles and can affect breathing and, in some cases, the heart. at 19 years old and I am now 20. I first noticed symptoms the beginning of 2019 and was diagnosed October 1, 2020. Towards the end of my senior year of high school, I noticed walking up the...

Genevieve’s Story
January 2022 My name is Geneviève, and I am 42 years old. I was diagnosed with Pompe disease at the age of 40. Here is my story. In my early thirties, I was skiing a lot with my 9-year-old daughter at that time. It was during this activity that my daughter pointed out...

Elizabeth’s Story
January 2022 I was diagnosed with LGMD when I was 12 years old. I had difficulty all my life with running, going upstairs, and doing sports, but no matter how hard things were I always pushed myself. I started working at 12 years old, started babysitting. At 16 years...

Margo’s Story
January 2022 This is Margot. She is almost two years old from Kentucky. She loves to swing, paint, and keep up with her 4-year-old brother. When she was born, there were no concerns. We went home and adjusted to our new life with baby number two. 8 days later, we...
Dwayne’s Story
December 2021 Hi everyone, my name is Dwayne; I am 53 years old. I was diagnosed with Late Onset Pompe disease (LOPD) in November 2018 when I was 50 years old. I live in Southern California with my wife and mother-in-law. I am a father to four sons. I also have a...

Haley’s Story
November 2021 My name is Haley Hayes. I am from Virginia. I am 16 years old and was diagnosed with infantile-onset Pompe DiseaseA severe form of Pompe disease that begins in infancy and often affects the heart and muscles. at 6 months old. I will be sharing my experience being a teenager living with Pompe and how it has affected me over the years. In the...

Caitlin’s Story
August 2021 My name is Caitlin Naldoza. I am 17 years old and was diagnosed with late onset Pompe Disease at 5 years old. I will be sharing my experience being a teenager living with Pompe and how it has affected me over the years. In middle school, that was when I...
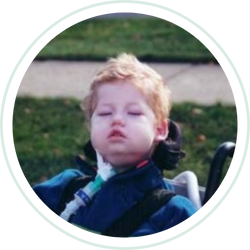
JOHN’S STORY
A year ago today [April 29, 2004] our son, John, passed away from Pompe Disease. He was four years old. Most of you will not recognize his name, but you all should know who he is. John was patient 101 – the first person to receive the CHO Enzyme Replacement Therapy...
WE NEED YOUR HELP!
To further research into Pompe Disease, as well as support Pompe patients around the world, private funds must be raised. If you are interested in learning more about Pompe Disease and would like to make a contribution in support of necessary research, please contact us at:
AMDA (Acid Maltase Deficiency Association)
P.O. Box 700248
San Antonio, Texas 78270-0248
Phone: 210-494-6144
Email: info@amda-pompe.org

Acid Maltase Deficiency Association
For the AMDA videos: All Rights reserved. No part of the AMDA's videos may be reproduced or transmitted in any form or by any means, electronic or mechanical, without the written permission of the copyright holder.
If you are interested in learning more about Pompe Disease and would like to make a contribution in support of necessary research, please contact us at:
info@amda-pompe.org
THE AMDA
PO Box 700248
San Antonio, Texas 78270 USA